
Abstract
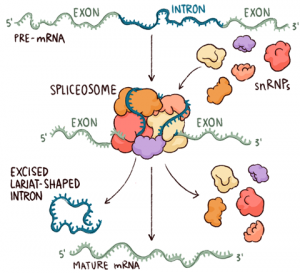
The spliceosome is a large RNA-protein complex that catalyzes the removal of introns from nuclear pre-mRNA. A wide range of biochemical and genetic studies show that the spliceosome comprises three major RNA protein subunits, the small nuclear ribonucleoprotein (snRNP) particles U1, U2, and [U4/U6.U5], and an additional group of splicing proteins. not snRNP. factors Rapid progress is being made in unravelling the interactions that take place between these factors during the splicing reaction.
The emerging picture of the spliceosome reveals a highly dynamic structure that assembles into pre-mRNA transcripts in a stepwise pathway and is organized, at least in part, by complex RNA base-pairing interactions between small nuclear RNAs (snRNAs) and the intron substrate. Many of these interactions can be detected in both mammalian and yeast spliceosomes, suggesting that the basic splicing mechanism is ancient and largely conserved during evolution.
What are spliceosomes?
Spliceosomes are huge multimegadalton ribonucleoprotein (RNP) complexes found in eukaryotic nuclei. They assemble into RNA polymerase II transcripts from which they extract RNA sequences called introns and splice flanking sequences called exons. This so-called pre-messenger RNA (pre-mRNA) splicing is an essential step in eukaryotic mRNA synthesis. Each human cell contains approximately 100,000 spliceosomes, which are responsible for removing more than 200,000 different intron sequences. Human cells contain two types of spliceosomes: the major spliceosome, which is responsible for removing 99.5% of introns, and the minor spliceosome, which removes the remaining 0.5%.
How did the various parts of the spliceosome get their names?
U snRNAs were originally discovered as abundant small uridine-rich RNA molecules present in mammalian nuclei and were initially numbered in order of their apparent abundance. U1, U2, U4, U5, U6, U11, and U12 were later found to be components of the splicesome. The U7 snRNA is required for processing the 3′ end of histone mRNA; the other abundant U snRNAs (U3, U8, U9, and U10) are all involved in ribosome biogenesis. U4atac and U6atac are much less abundant than other spliceosomal snRNAs, so they were only discovered and named when it was realized that there must be other snRNAs that recognize the minor intron class.
The first and last two DNA nucleotides of minor introns are usually AT and AC, respectively, hence the names U4atac and U6atac. Many spliceosomal proteins have PRP names, e.g. Prp2, Prp5, Prp8, etc. In yeast, mutations in these genes lead to “mRNA pre-processing” defects. Confusingly, orthologous genes may have different PRP names in Saccharomyces cerevisiae and Schizosaccharomyces pombe because the original mutational screens were done around the same time and a unified naming system has not yet been devised.
Other core splicing proteins include CWC (complexed with CDC5), CWF (complexed with CDC five), SPF (Pichia farinosa killer toxin sensitivity), SYF (synthetic lethal with cdcforty). Complex nineteen (NTC) is a large protein-only subcomplex named for its most abundant component, Prp19, while another small protein-only complex known as NTR (related to complex nineteen) contains factors involved in spliceosome disassembly. Some of the major spliceosomal proteins were first discovered in invertebrates.
The seven Sm proteins, which form a ring surrounding a specific binding site on almost all spliceosomal snRNAs, were named after the patient (Smith) with whose autoimmune antibodies they react. A similar set of proteins (Lsm, for “Sm-like”) was later found to surround the U6 and U6atac snRNAs, the only two spliceosomal snRNAs that lacked a consensus Sm binding site. Two additional large classes of metazoan splicing factors are the hnRNP proteins, so named because they are found associated with heterogeneous nuclear RNA (hnRNA), and the SR proteins, named for a carboxy-terminal domain rich in arginine-serine dipeptides ( RS).
How does the spliceosome do its job?
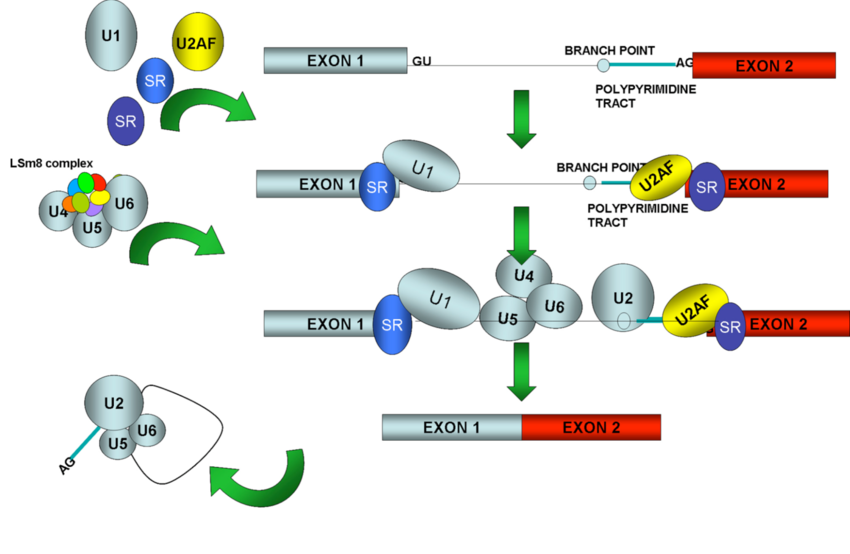
Spliceosomes must remove non-coding introns from precursor transcripts and rejoin flanking exons to create mature spliced mRNAs. To do so, splicing machinery assembles step by step at the ends of introns, with U1 snRNP recognizing the start of an intron (5′ splice site, the donor site) and U2 snRNP recognizing a feature (the donor site). branch) at the other end in the vicinity of the 3′ splice site (acceptor site).
After numerous structural rearrangements involving both the addition of new components and the expulsion of many others, splicing occurs in two chemical steps: first, cleavage at the 5′ splice site along with the formation of a lariat structure. in which the first nucleotide of the intron is linked via a 2′–5′ phosphodiester bond to the adenosine branch site; and second, ligation of the two exons, along with cleavage at the 3′ splice site. The spliceosome then disassembles from the excised intron, which subsequently debranches and degrades.
How do spliceosomes affect gene expression?
Because the vast majority of protein-coding genes in humans contain introns (usually 9 or 10, but some have more than 100!), splicing is an essential step in gene expression. High-throughput sequencing has now revealed that ~95% of human genes are also subject to alternative splicing, allowing the synthesis of many different mRNAs from a single DNA gene. By encoding alternative protein isoforms or harbouring different regulatory sequences in their untranslated regions, alternatively spliced mRNAs greatly enhance biological complexity.
The act of splicing itself also has important consequences for gene expression beyond intron removal. By stably depositing proteins that accompany mRNPs in the cytoplasm (e.g., the exon-joining complex, EJC) into exons, splicing can affect subcellular localization, translational efficiency, and decay kinetics of mRNP. mRNA. In particular, mRNA decay driven by the location of EJC relative to the stop codon is a crucial mediator of cellular protein abundance.
Are spliceosomes associated with any disease?
Many human diseases are caused by the misplacing of a single gene or by the dysregulation of the entire spliceosome. About 35% of human genetic disorders are caused by a mutation that disrupts the splicing of a single gene. Such mutations can add/delete a single splice site (eg, α or β thalassemia) or change the balance of alternative splicing by affecting the inclusion/exclusion of a cassette exon (eg, frontotemporal dementia driven by a misplacing of tau). Some misplacing events generate an isoform of mRNA that is subject to rapid degradation.
Single point mutations affecting splicing can result in large changes in both protein structure and protein abundance. Other diseases are caused by mutations in the splicing proteins themselves, affecting the splicing of many transcripts. For example, mutations in several core splicing proteins (eg, Prp8, Prp3, Prp31, and Brr2) have been shown to cause autosomal dominant retinitis pigmentosa. Mutations in splicing factor 3B subunit 1 (SF3B1) and U2 helper factor 35 (U2AF35) are frequently associated with chronic lymphocytic leukaemia and myelodysplasia. Other types of cancer are associated with dysregulation of splicing factor levels. Therefore, the spliceosome has recently emerged as a new target for the development of new cancer therapies.
What is left to explore?
Due to its highly dynamic and complex nature, an atomic-level structure of the spliceosome remains an elusive goal. However, much progress has recently been made by crystallizing subsets of spliceosomal components, including the U1 and U4 snRNPs and the central core protein Prp8. Other important questions concern the exact molecular mechanisms by which spliceosomes achieve high splicing precision while allowing flexibility in splice site choice to enable alternative splicing. To answer these questions, new tools such as single-molecule microscopy, bioinformatics, and high-throughput methods for determining protein-protein, protein-RNA, and RNA-RNA interaction dynamics are increasingly being developed and applied.